测序使传统的序列组装变得简单,同时保持您控制。
轻松修剪序列,以创建质量差的数据。您甚至可以维护修剪标准库,使生活更轻松。
通过直观的控制,您可以为您的数据选择最佳算法,包括”组装到参考”。如果您正在处理来自不同源的多个示例,则使用”按名称组装”来自动执行程序集。
使用工具来帮助您查找和处理歧义、检查异质和移动数据,对数据进行编辑从未如此简单。
使用置信度值来帮助修剪数据、质量检查和 SNP 检测,以提高结果的质量。
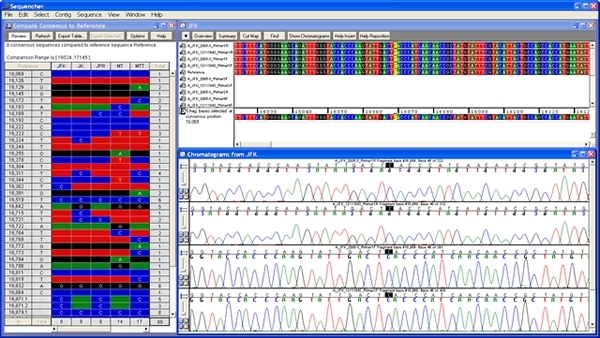
测序器为您提供了DNA序列编辑工具,您需要知道序列是绝对正确的。您可以一次查看一个序列的色谱图数据,或以向前和反向方向查看多个对齐的色谱图。
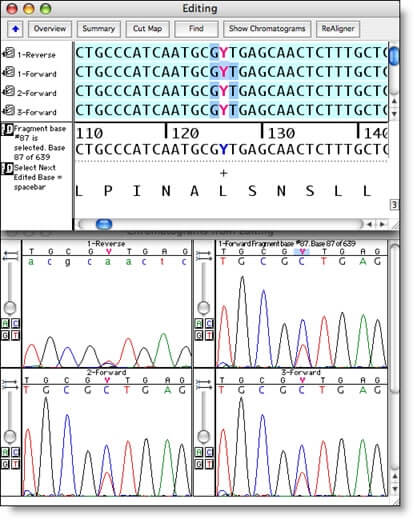
快速轻松地滚动查看对齐的数据,或者您可以使用 Sequencher 的选择工具突出显示差异或低质量区域。
有关测序器的DNA序列选择和编辑工具的信息,请查看测序器指南。
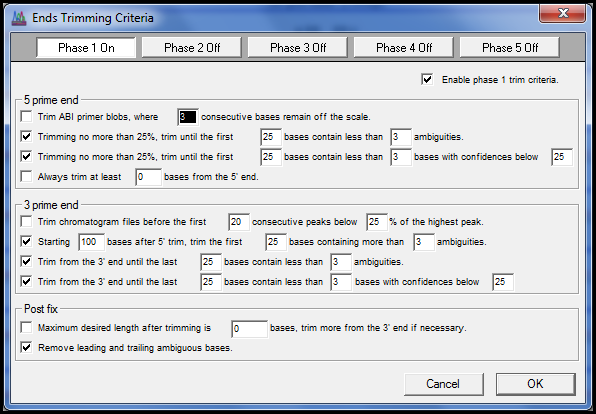
自动DNA测序器偶尔会产生质量差的读取,特别是在测序底转位点附近,并且接近较长序列运行的末尾。来自DNA库的克隆序列通常包含矢量序列、聚体A尾部或其他无关序列。内子和底因序列经常侧翼放大外向序列。除非通过修剪删除,否则任何这些伪影都会扭曲序列装配体和下游序列分析。
Sequencher 提供简单易用但功能强大的工具,可帮助您修剪质量差或模棱两可的数据:
修剪端点从测序片段的末端删除误导性数据。
修剪矢量删除污染序列末端的特定于序列的数据。
修剪到参考消除了超出组装的参考序列的序列的末端。
在执行修剪之前,Sequencher 会显示建议的修剪的图形表示形式,这允许您进一步优化条件。

如果要在修剪序列的任一端或两端还原一组基点,因为修剪过于严格或想要提高覆盖范围,则批处理还原修剪结束允许您这样做。只需单击几下,即可将基座还原到几千个序列或几千个序列,并更好地控制序列修剪。
Sequencher 的直观控制允许您设置序列装配参数并在几秒钟内调整它们,从而快速准确地组装 DNA 片段。测序器将自动比较前进方向和反向补数方向,以组装最佳连音,因此您可以组装 DNA 序列,无论方向如何。
将测序器的多功能装配工具应用于

您可以在”装配策略教程”中找到有关这些工具的更多信息。
参考序列是一个强大的功能,是测序和序列分析的许多方面的核心。无论您是SNP狩猎,从事法医,植物遗传学研究,医学遗传学或人口研究,你都将希望使用参考序列功能。
以 GenBank 格式导入序列,其特征表将应用于序列,并将它标记为参考序列。组合到参考序列意味着您可以比较您的读取与原型参考序列。如果您正在处理来自不同来源的多个示例,甚至可以使用”按名称组装”来自动执行工作。
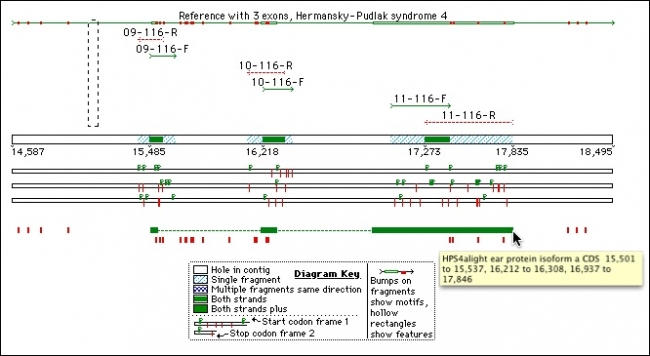
您甚至可以使用参考序列来指导在感兴趣区域之外删除序列或填补序列覆盖率中的空白。
使用方差表及其强大的报表可视化结果。
自4.9 版以来,Clustal [1] 一直是序列插件系列的一部分。它是一种广泛使用的多序列对齐程序,它通过确定一组序列上的所有成对对齐来工作,然后构造一个树突图,按近似相似性对序列进行分组,最后使用脱光图作为参考执行对齐。您可以使用 Clustal 直接从”排序器”项目窗口对齐序列。从一系列参数中选择以控制对齐过程。
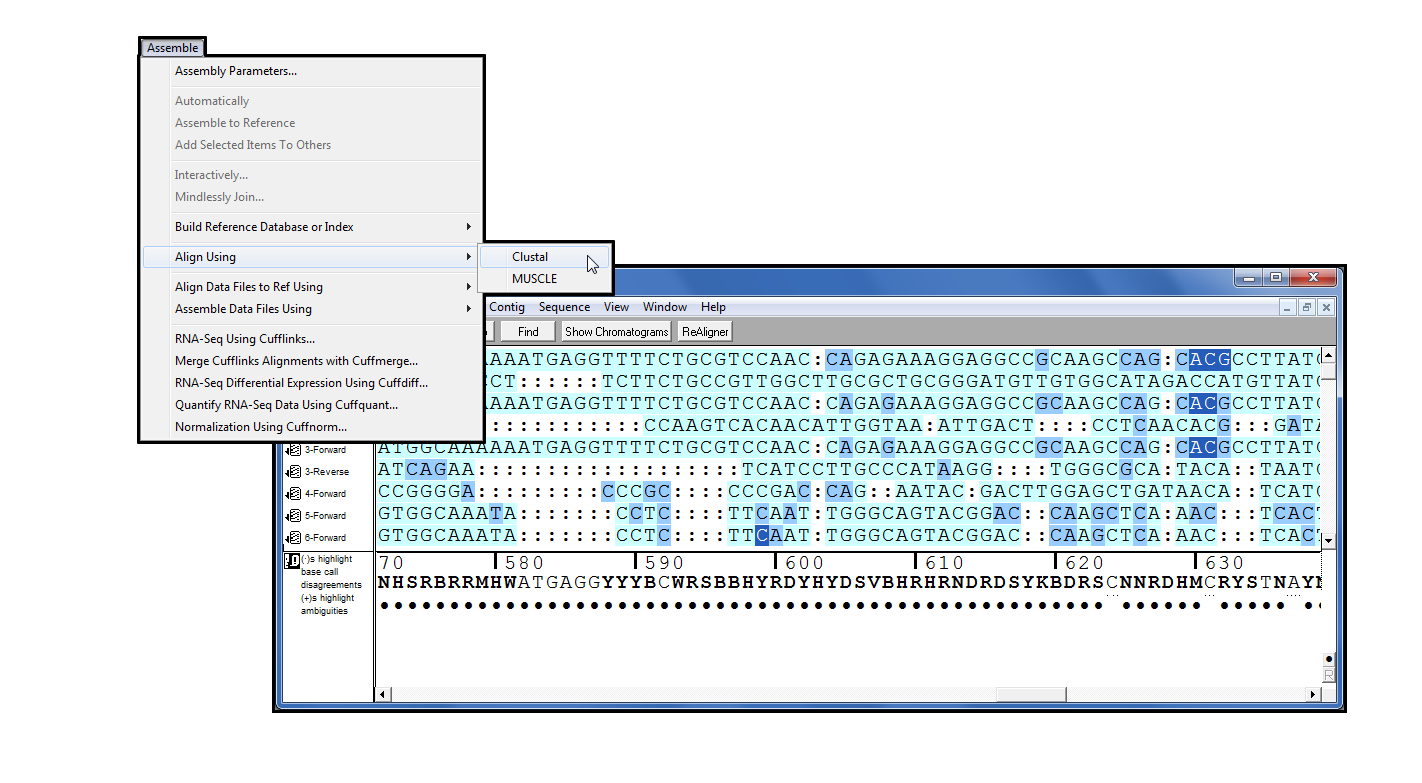
一旦对齐完成,您将在测序器中看到结果作为连体或连体,您可以接受各种分析。
通过将 Clustal 与 Sequenceher 的按名称组装功能的功能相结合,以对齐来自不同源的多个序列,加快您的 Clustal 对齐速度。
最后,以各种不同格式导出结果,如 MSF、Phylip、NEXUS 和 FastA,用于其他程序,或者只需创建共识并导出即可。
肌肉
肌肉[2], 多序列对齐 (MSA) 程序, 加入序列 5.1 系列插件.它加入了Clustal,成为测序器DNA-Seq工具中的第二个MSA程序。它拥有速度和精度,与其他多序列对齐程序相比非常有利[3]。
据说,MUSCLE 在对齐过程中有四个主要步骤。第一步使用 k-mer 聚类构造序列对之间的距离矩阵,然后将其转换为树。第二步使用此树引导渐进式对齐。在最后两个步骤中,MUSCLE 算法尝试许多不同的方法,以查看是否可能改进树,从而改进多个对齐方式。
一旦过程完成,肌肉已经建立了对齐,你会看到结果作为一个连在序列。使用 Sequencher 的工具对对齐进行注释或导出对齐方式,并放入特殊的植物遗传学程序中。
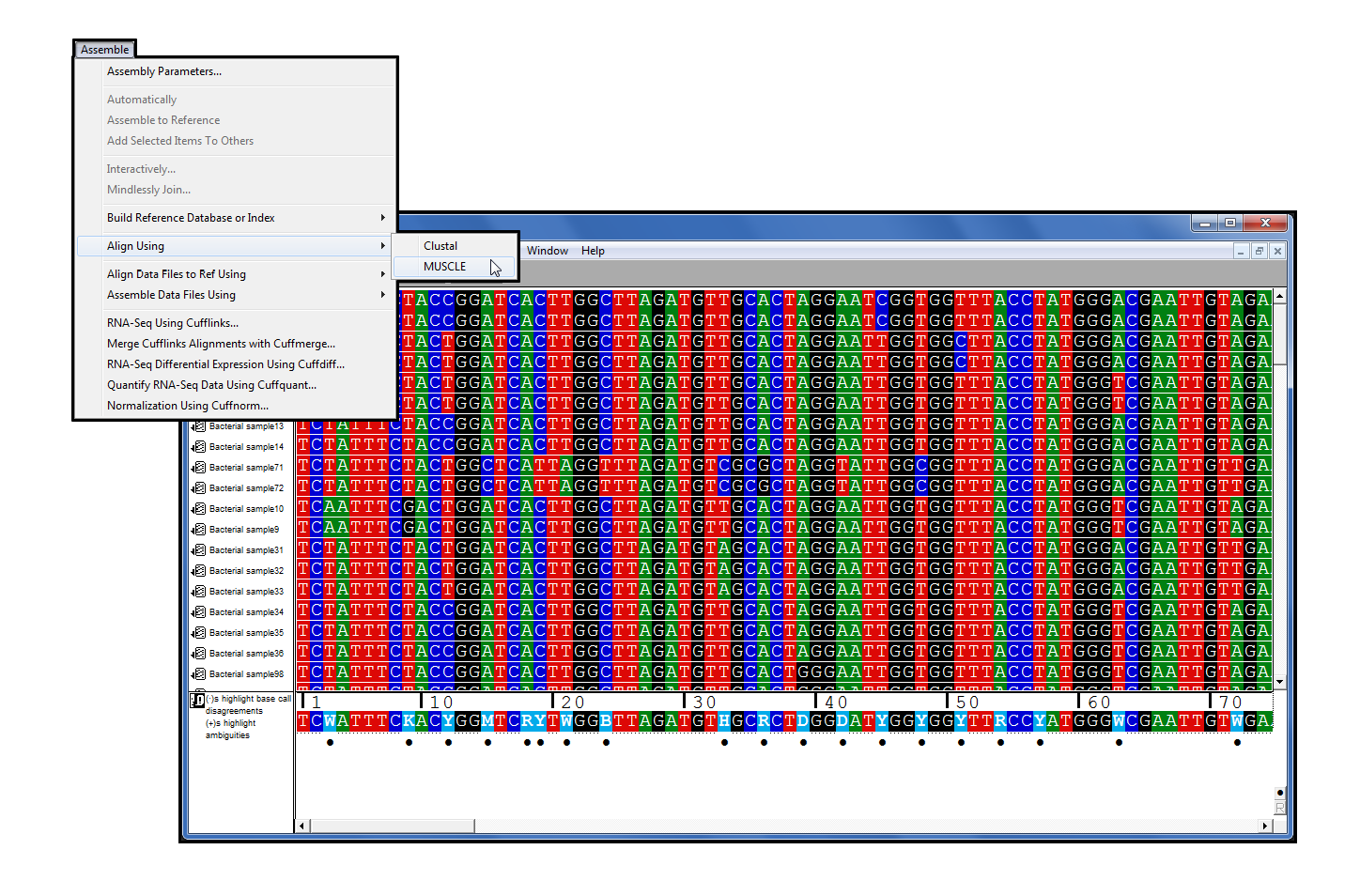
MUSCLE 是一个命令行程序,这意味着通常您将通过终端应用程序使用此程序。排序器允许您访问 MUSCLE 的电源,而不会遇到学习使用 UNIX 命令行的问题。
[1] 陈娜 R, 苏加瓦拉 H, 科伊克 T, 洛佩兹 R, 吉布森 Tj, 希金斯 Dg, 汤普森 Jd (2003).”与 Clustal 系列程序的多个序列对齐”。核酸 31 (13): 3497-3500
[2]核酸。2004年3月19日;32(5):1792-7。肌肉:具有高精度和高吞吐量的多个序列对齐。埃德加·RC.
[3] BMC 生物信息学。2004 8月 19;5:113.肌肉:一种多序列对齐方法,可降低时间和空间的复杂性。埃德加·RC.
测序器提供了一套丰富的工具,用于生成DNA序列的线性限制图。按频率、悬伸性质或识别序列长度过滤酶选择。
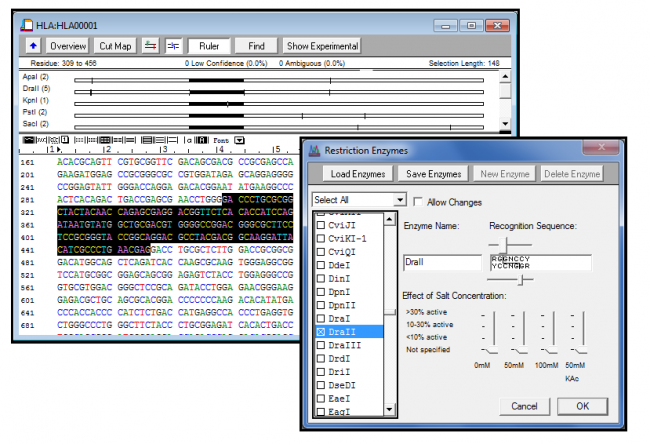
您还可以指定特定的矢量和多链接序列,以帮助您设置克隆策略。
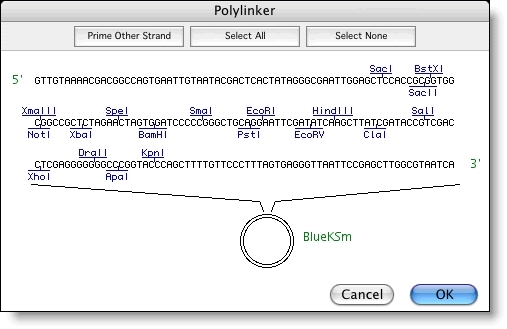
排序程序在 Project 窗口、序列编辑器和”序列获取信息”窗口中显示置信度和汇总置信度信息(如果 DNA 序列文件中可用),以便轻松监视数据的质量。
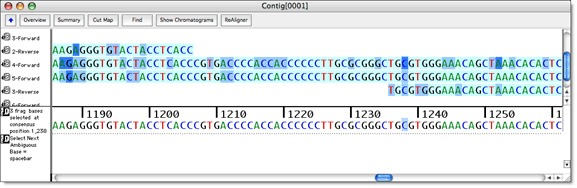
您甚至可以为置信值指定截止范围,并按颜色代码查看这些范围。
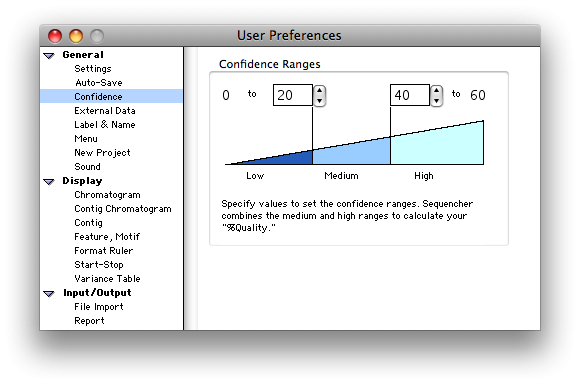
有关 Sequencher 如何处理质量分数的信息,请参阅质量分数教程。
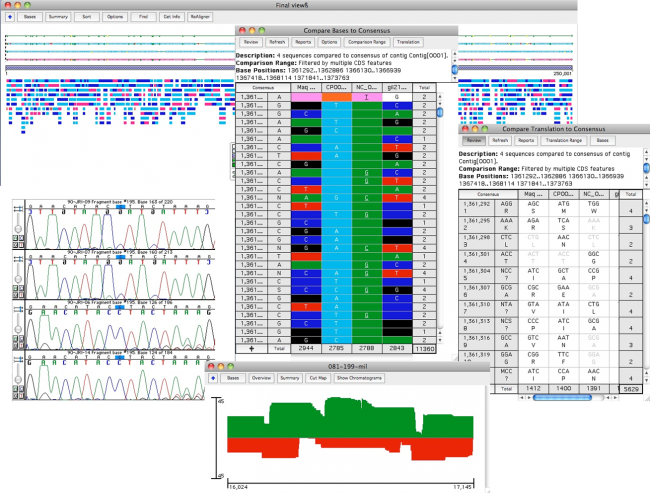
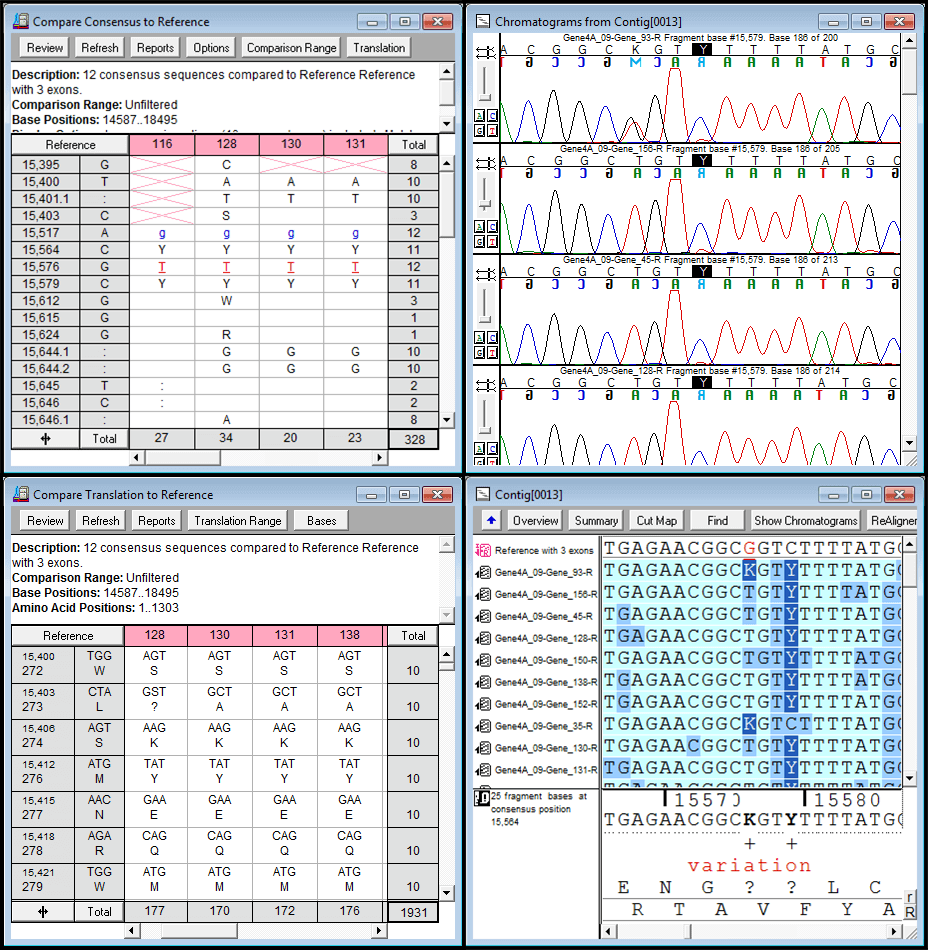
Sequencher 有几个强大的工具,可帮助您检测DNA序列中的突变和SNPs。您可以使用 Sequencher 在一组序列之间比较序列对齐,或将 1 个或更多序列与参考序列进行比较。测序器的呼叫二次峰值…很容易控制定义异质的字符串性。
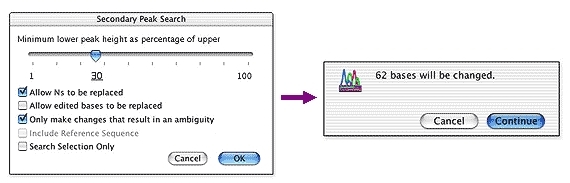
映射DNA组装概述中所有异质异质的各异质体的位置。
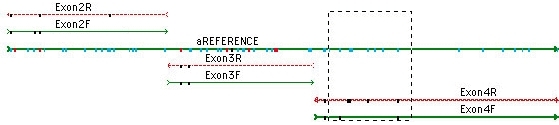
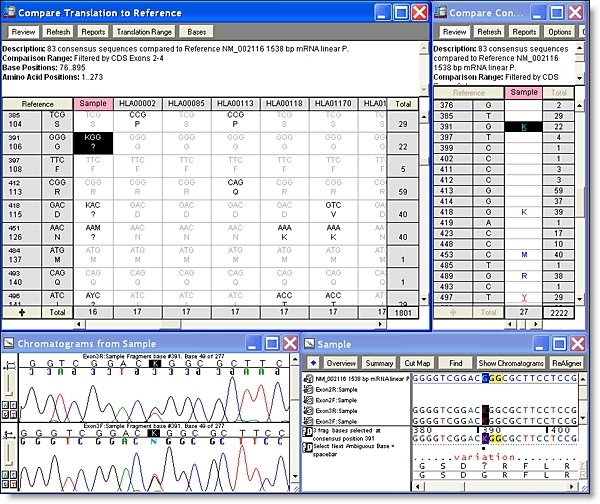
你可以从一个异质戈特导航到下一个;只需单击”基地”视图中的空格键。查看共识和参考序列的蛋白质转换,以在共识下方。参考序列可确保 SNP 的编号从一个 DNA 组件到下一个 DNA 程序集保持一致。

有关使用测序查找SNP的更多详细信息,请参阅有关SNP狩猎的教程和有关SNP狩猎的教程。
Sequencher 批处理您的DNA序列数据的方式是透明、用户可定义和可恢复的,而测序器绝不会为了自动化而损害科学结论的有效性。排序器始终为您提供序列编辑的最终选择。
排序器始终维护数据的两个副本,即编辑的数据和原始导入的数据。将”还原到实验数据”命令应用于项目中的序列选择或序列中的一部分基础时,可以撤消编辑的左右部分。
“按名称组合”工具允许您选择片段名称的一部分作为共享标识符或”程序集句柄”。然后,排序器会自动进行选择并命名连结。排序器甚至支持正则表达式匹配以设置唯一的 ID!

例如,只需单击一个按钮,即可将 90 个文件、45 对前进和反向序列转换为 45 个连音,根据患者 ID 命名。序列装配体参数中的更改会重新组合片段,因此您可以根据克隆 ID、日期、Primer 或在序列名称中记录的任何其他特征对连体进行组合。
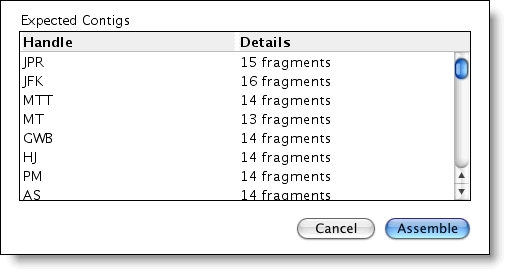
如果您进行大量测序,并且有许多样本使用一组标准测序底像完成,那么按名称组装尤其有用。按名称组装的一些其他应用程序可以包括:
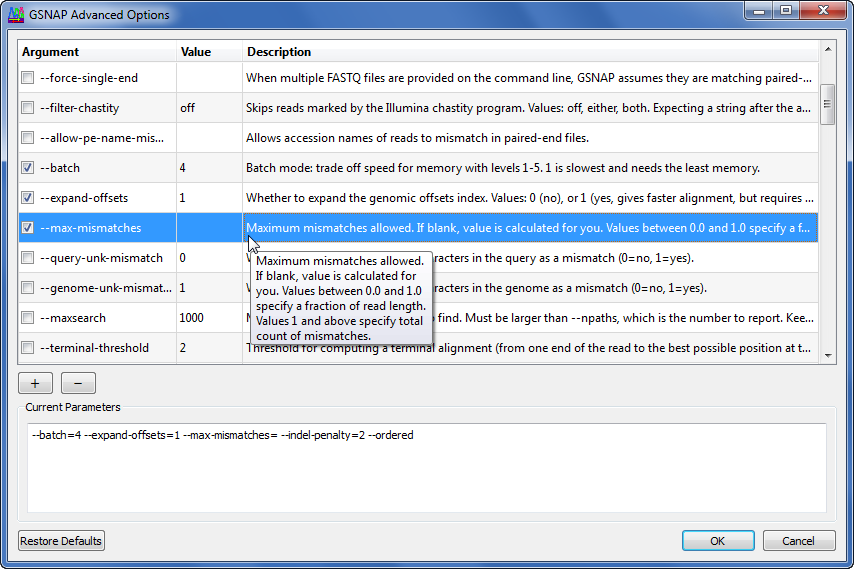
Gene Codes 致力于在同行评审期刊上开发和发布各种对齐和装配算法,但非工程师可以访问这些算法。例如,GSNAP(Tom Wu,Genentech, Inc.)被公认为正确识别拼接结的最佳算法之一。生物信息学专家可能会这样调用程序:

如果我们有一些工具来帮助我们了解所有可用的标志和参数是什么,以及它们如何影响程序,那么我们大多数人可以更高效地工作。Sequencher 界面可帮助您选择选项、设置值,并了解可用功能以及说明和工具提示。上面的命令行中的相同选项在排序器中按以下方式设置:
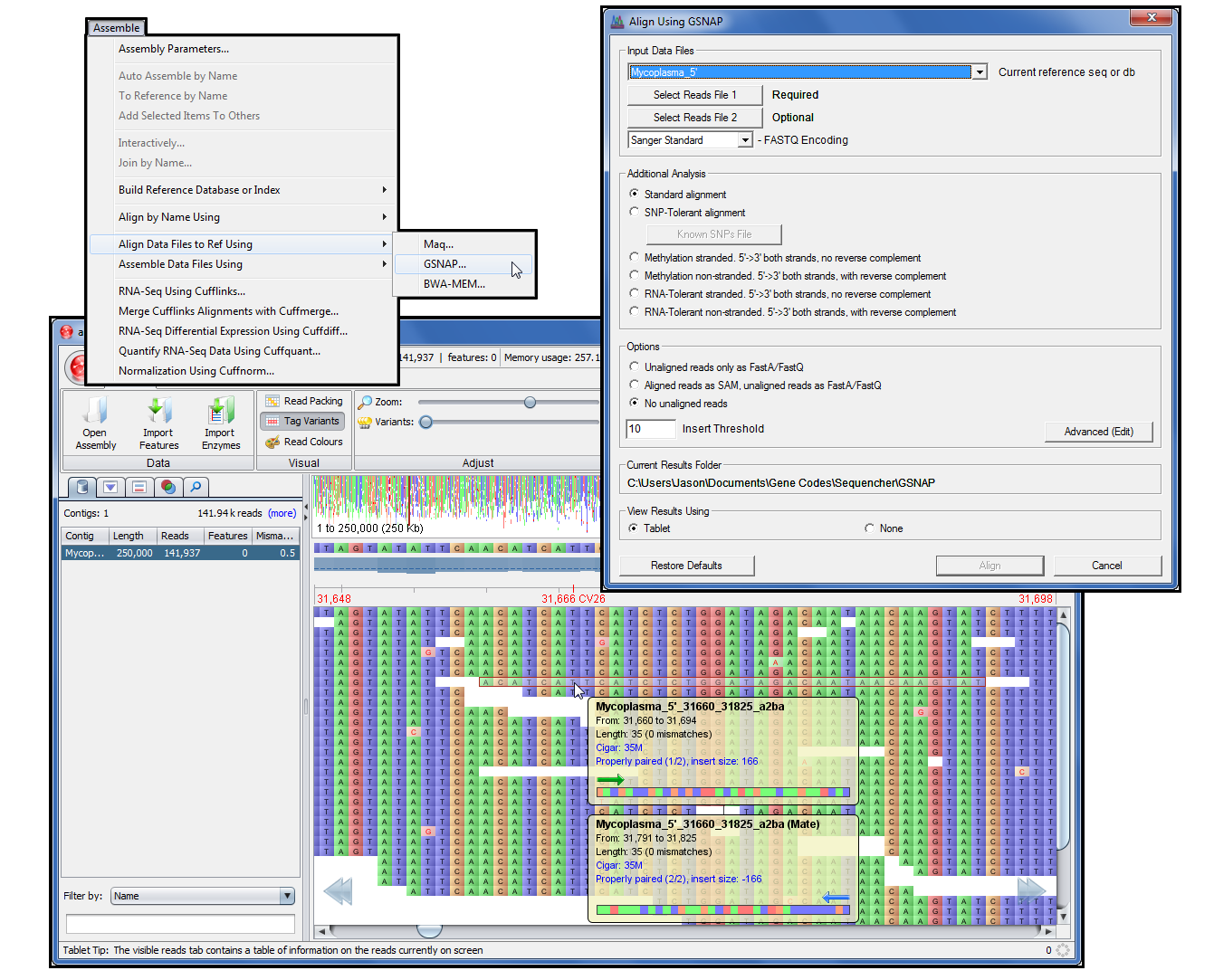
只需几个步骤执行SNP分析、甲基化分析或RNA A-G容差对准。
在平板电脑中查看结果。
BWA、天鹅绒、Maq、GSNAP 和平板电脑只是测序器的开始。如果要对混合群体进行测序,则将参考引导对齐功能与 de novo 装配体相结合。将读取与引用对齐并捕获未对齐的读取,以进行进一步的基于引用的对齐或 de novo 装配。
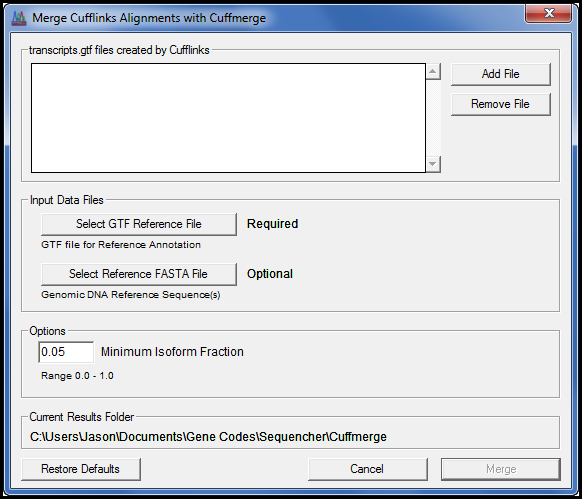
通过添加 Cufflink 套件,您可以使用我们为所有 NGS 算法开发的相同接口样式执行 RNA-Seq 差分表达。
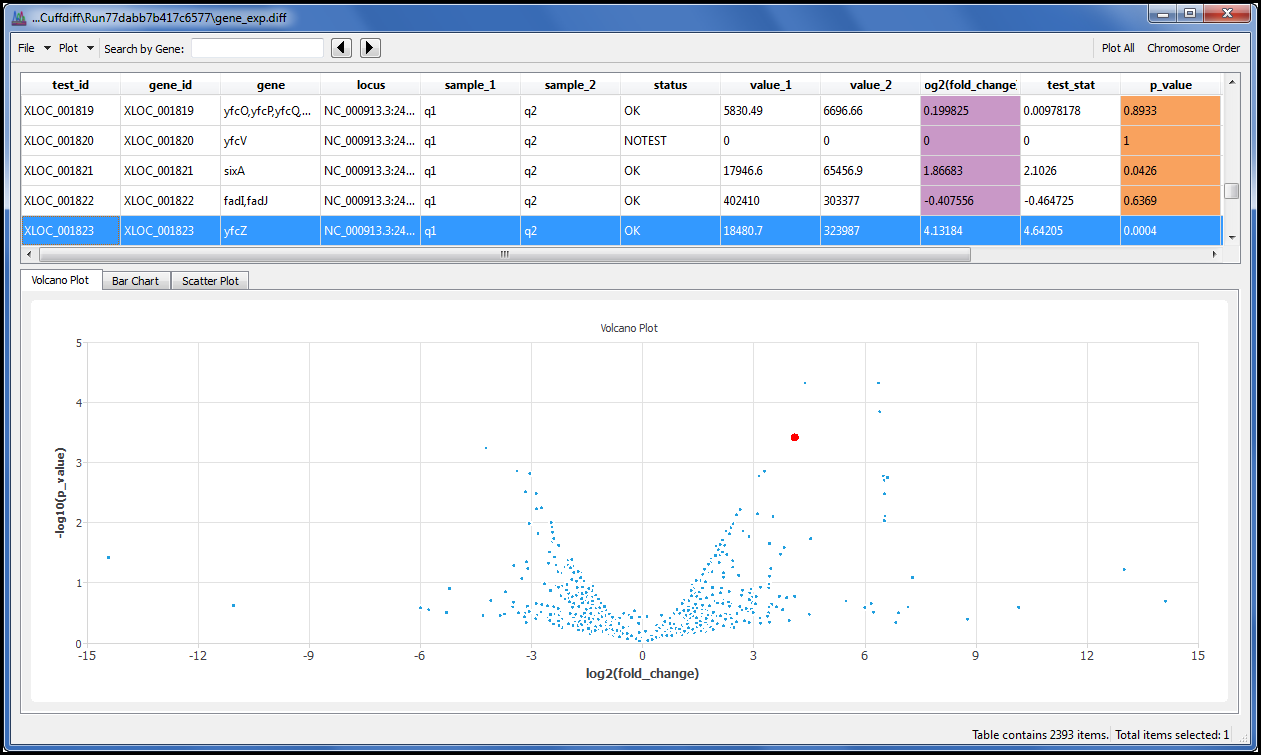
我们还添加了预期看到的图表,如火山图,因此无需使用命令行即可查看结果。因为我们知道你有你最喜欢的对齐器,我们不坚持你使用我们的。只要对齐器创建 SAM 或 BAM 作为输出,您就可以使用最广为人知的 RNA-Seq 工具之一来探索您的数据。
在开始对齐数据之前,请稍等片刻考虑。难道你不想知道你的数据有多好吗?好了,现在你可以与 FastQ 质量报告。我们已经将流行的 FastQC 程序集成到了测序器中。从序列 > 分析 > FastQ 质量报告启动,您可以获得多达 12 个不同指标的结果。从”按基本序列质量”到”Kmer 内容”,从”序列重复级别”到”代表性序列”,结果都配有易于理解的红绿灯系统以及更详细的图形。
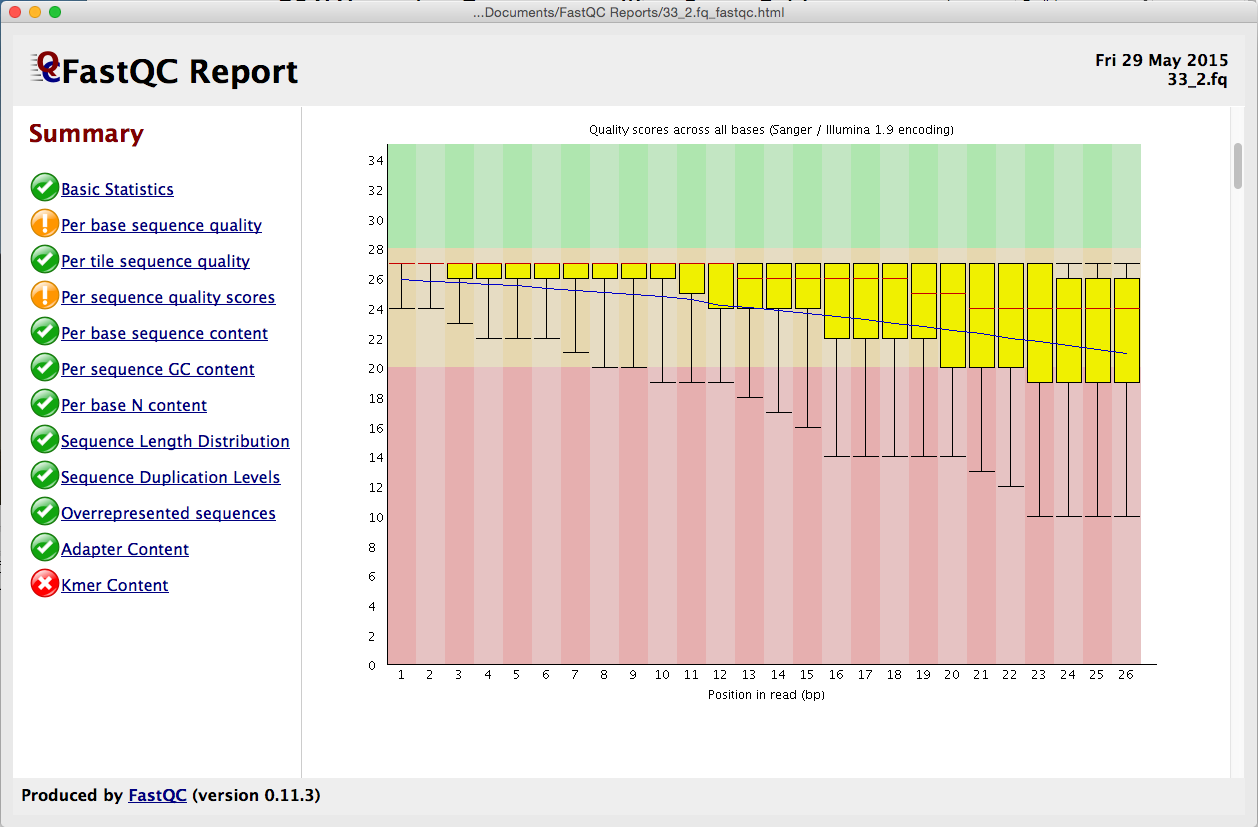
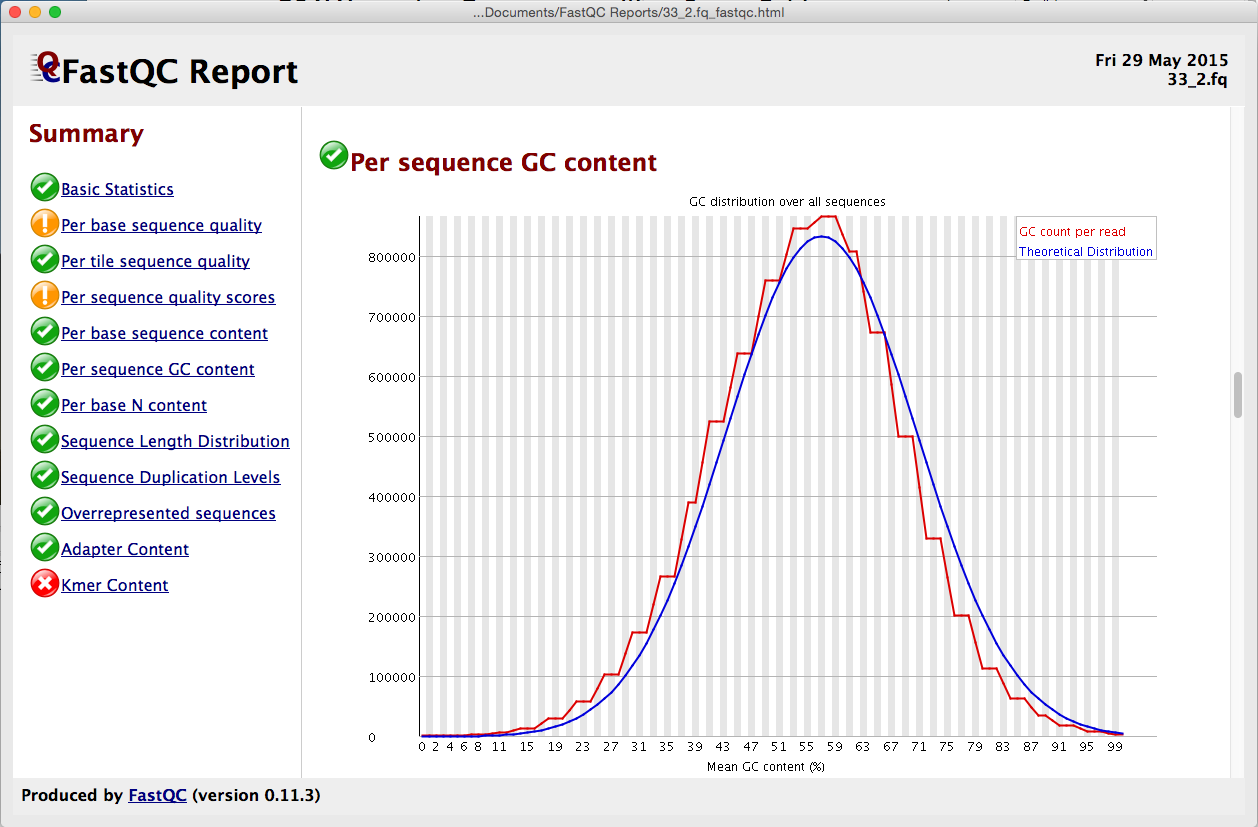
每个报表都在其自己的窗口中显示,因此您可以并排查看它们。使用报告在组装之前监视个人数据集的质量,或监视一段时间的质量。有关 FastQ 质量控制报告的信息,请查看 FastQ 质量控制报告教程。
RNA-Seq实验为蛋白质编码记录的研究带来了新的理解和知识,无论是来自不同时间点的正常组织,还是正常和疾病状态之间的正常组织。Sequencher 插件系列的最新成员是 Cufflinks 套件[1],这是一系列专为研究 RNA-Seq NGS 数据而设计的程序。入门再简单了。
使用您最喜爱的对齐器将 RNA-Seq NGS 数据与参考序列对齐,然后获取生成的 SAM 或 BAM 文件和参考 GTF 文件以开始使用 Cufflink。
重点是使用袖扣、袖扣、袖口四步过程的差分表达式,最后将您的数据显示为表和绘图。Cufflinks 套件命令行程序都可以使用我们易于使用的图形界面访问。对于最终控制,您还可以通过简单点和单击”高级(编辑)”对话框访问所有高级命令行函数。
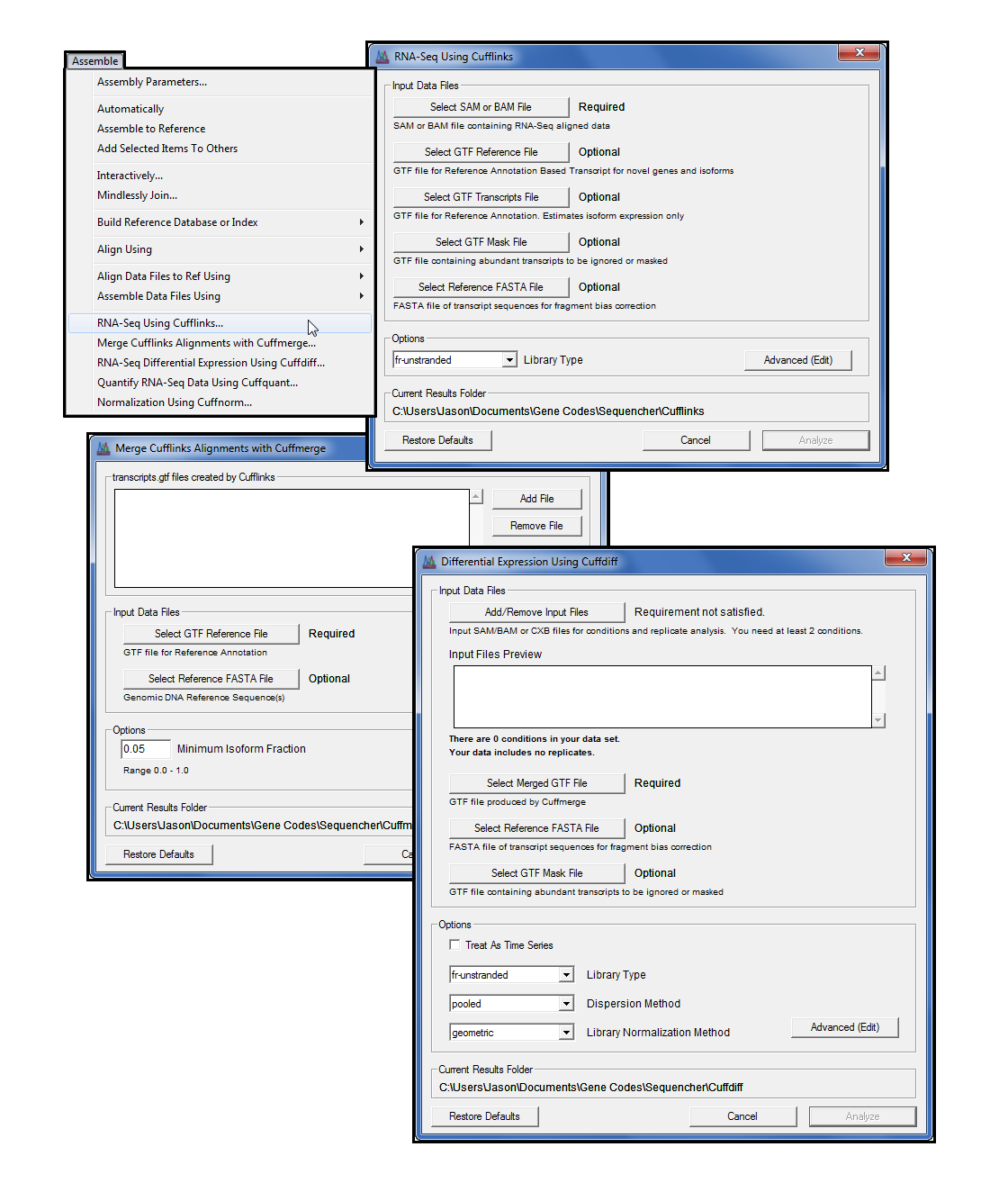
您可以运行分析,并在 Sequencher 中以绘图查看结果。通常,差异表达式要求您安装和使用命令行中的 R 统计编程语言。
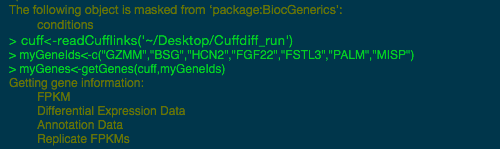
我们为您提供一个菜单命令,允许您使用火山图、散点图和条形图显示结果。每个点或条都链接到其基础数据,因此您可以单击您感兴趣的点或柱,并详细浏览结果。
Sequencher 中的袖扣套件现在包括袖口和袖口。如果您没有研究不同表达的基因,但仍需要规范化的结果,请使用袖口和袖口后袖扣步骤。如果需要减少计算机上的计算负载,可以在”袖口”和”袖口”步骤之后使用 Cuffquant 来量化数据集中的读取。然后,当您准备就绪时,使用 Cuffdiff 完成差分分析。你会发现这个步骤要快得多,因为那部分工作已经完成。
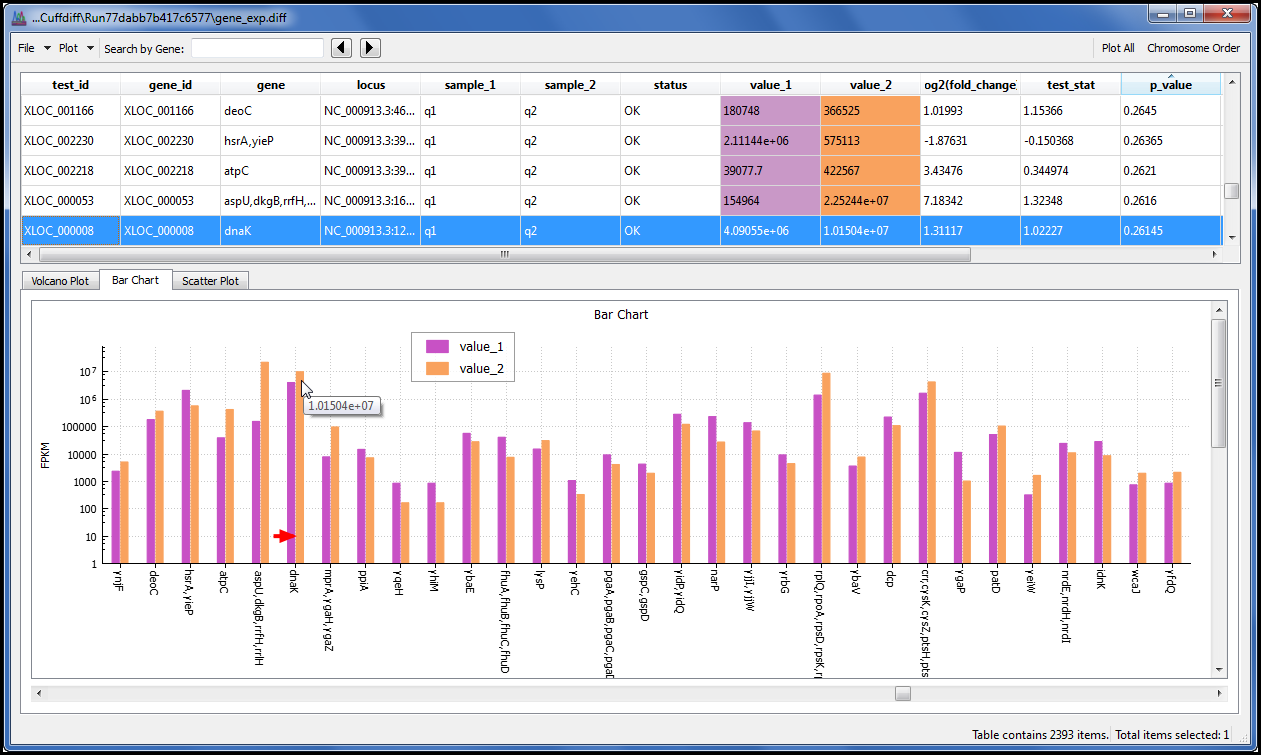
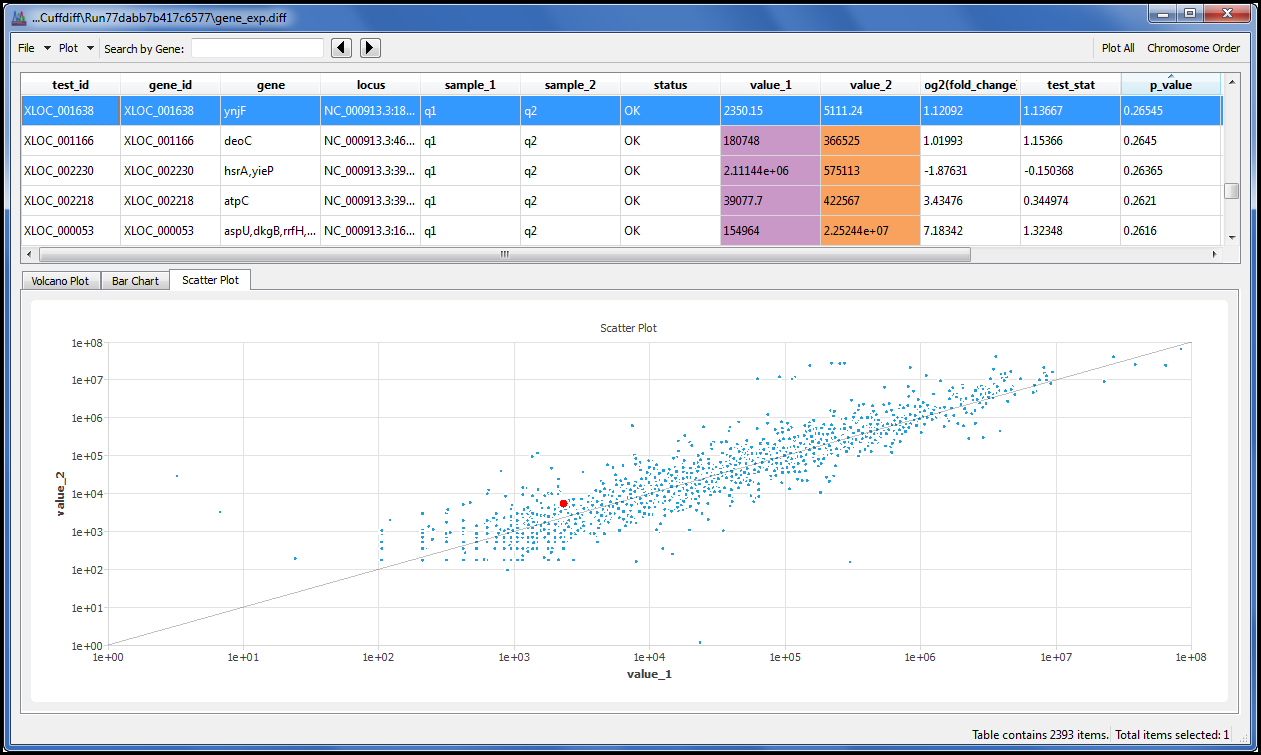
按基因名称或基因 ID 搜索数据表,快速找到您感兴趣的数据。
排序和筛选数据,仅绘制您感兴趣的内容,然后以 PNG 格式导出图形以包含在演示文稿或报告中。
有关RNA-Seq的信息,请点击此处查看RNA-Seq教程。
[1] RNA-Seq 的转录和定量揭示了细胞分化过程中未注释的转录和等构交换
科尔 · 特拉普内尔、布赖恩 · 威廉姆斯、乔治 · 佩尔塔亚、阿里 · 莫塔扎维、戈登 · 宽、玛丽克 · 范巴伦、史蒂文 · 萨尔茨伯格、芭芭拉 · J · 特和 · 利尔 · 帕赫特
自然生物技术 28, 511+515 (2010)
Velvet (1)加入了版本 5.1 中不断增长的 Sequencher 插件系列。Velvet 是众所周知的德诺沃汇编器,它不需要参考序列。虽然我们谈论 Velvet ,它实际上包括两个程序。Velveth 准备数据集,而 Velvetg 使用名为 de Bruijn 图形的方法执行装配步骤。Velvetg 构建了连连,甚至会尝试脚手架的连连,而不是完全走到一起自己。
Velvet 可以使用多路复用 ID 数据执行 de novo 程序集。排序器会自动将数据按条形码划分为单独的文件(bin),然后对齐它们并将其结果放入单独的结果文件夹中。然后可以在平板电脑浏览器中查看结果。每个连结的一致顺序将显示在项目窗口中。
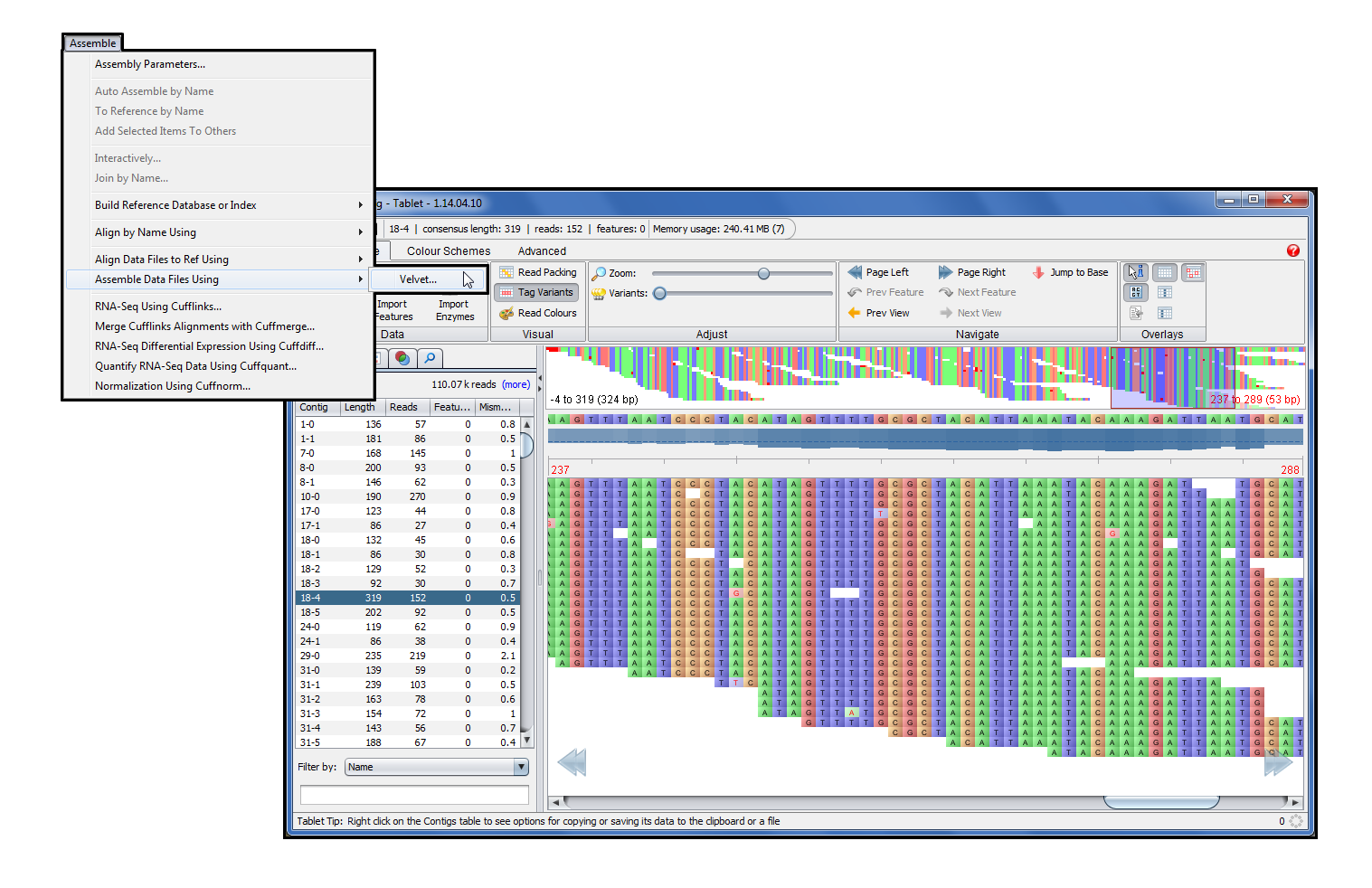
Velvet 被写入运行的命令行。Sequencher 通过只需单击几下即可启动程序集运行,即可保护您免受命令行的攻击。电源用户和新手仍然可以访问命令行参数来添加或更改参数,我们输入了一个新的 GUI,让您访问和利用这些参数。
如果您想了解有关使用 NGS 数据的 Velvet 和 de novo 程序集,请查看 De Novo 装配教程。
(1)Velvet:使用de Bruijn图形的 de novo 短读程序集算法。泽比诺和E.伯尼基因组研究18:821-829
对齐者花费一定的时间对参考序列进行索引,以加快整体对齐速度。现在,您可以创建并保留这些索引 (BWA) 或数据库 (GSNAP),以便当您想要尝试不同的参数以改善对齐或拼接放置时,速度会更快。
索引和数据库文件是跨平台的,因此您可以在不同类型的计算机上与协作者共享这些文件。创建新索引或数据库时,它将自动显示在该对齐器的可用引用序列列表中,随时可以使用。排序她记得它在哪里, 所以你不必。
来自预处理引用的文件可能会变得很大。当您不再使用时,只需使用外部数据浏览器单击一下即可将其删除。您将节省磁盘空间,如果需要再次索引相同的参考序列,则排序器可以轻松地重新创建它。
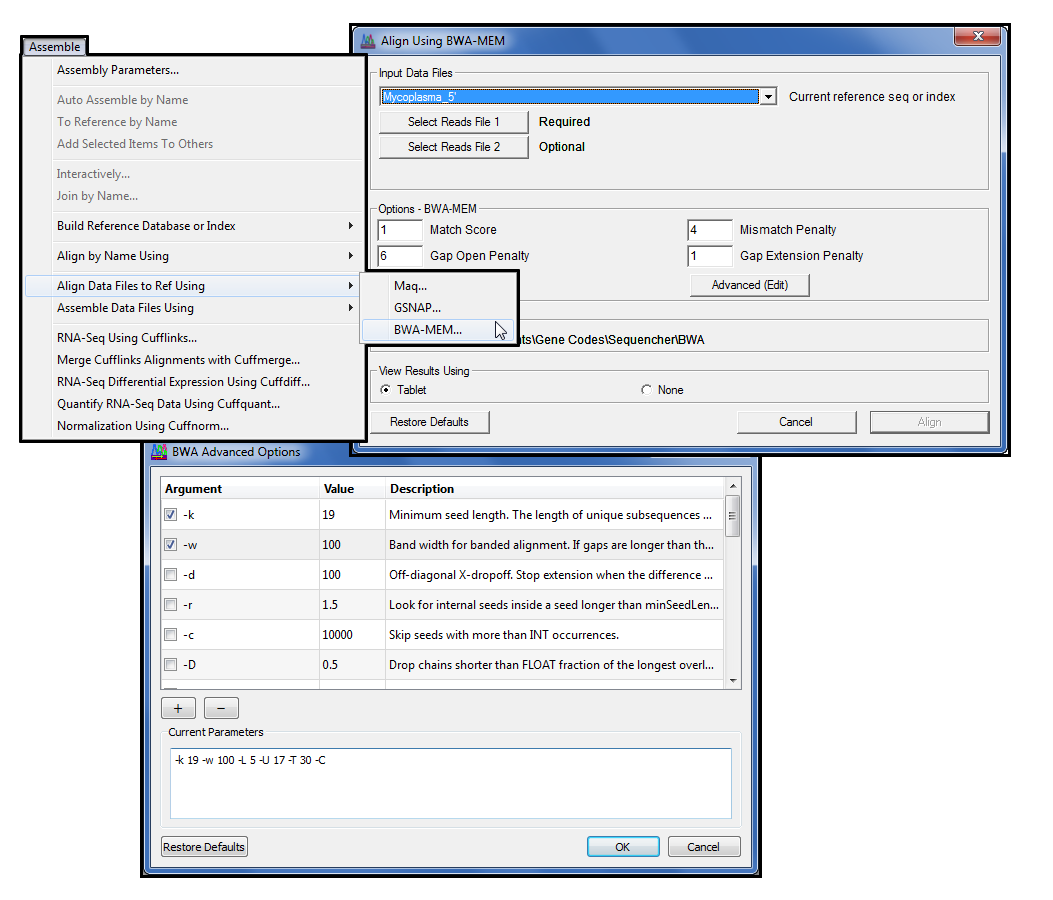
GSNAP(2)加入了版本 5.0 中的测序插件系列。GSNAP 使用高效的方法通过压缩参考序列进行基于参考的对齐。这使得 GSNAP 在执行下一代测序时更快、更容易。
GSNAP 旨在预制基于参考的照明-Solexa 或 Sanger 标准数据对齐,这些数据是成对端和未配对的。序列的长度不是 GSNAP 的问题,因为它能够轻松对齐非常短的数据长度到任意长的数据长度。
GSNAP 现在可以使用多路复用 ID (MID) 数据,从而进一步提高效率。现在,您拥有 Sequencher 的用户友好界面,允许您利用 GSNAP 的电源与您的 MID 数据。排序器会自动将数据按条形码划分为单独的文件,使用 GSNAP 将它们与引用对齐,并将结果放入单独的结果文件夹中。
与 GSNAP 的任何对齐结果都可以在流行的平板电脑浏览器中查看。
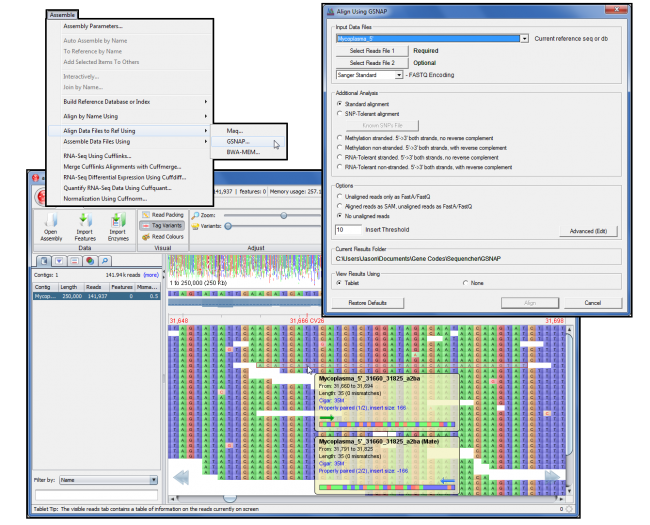
对于希望更好地控制 GSNAP 命令行功能的电源用户,按”高级”(编辑)打开高级 GSNAP 选项对话,您可以在其中配置特定的命令行参数。
Maq(3)加入了版本 5.0 中的序列插件系列。流行的 Maq 算法将单端和配对端的下一代数据与参考序列对齐。最初设计用于对齐 Illumina-Solexa 数据,它将对齐任何非常短的读取数据(63 个基或更少)。引用序列可能以 FastA 或 GenBank 文件的形式出现。Maq 使用二进制格式压缩引用并读取文件。最重要的是,Maq 只需很少的 RAM 才能运行,这使得执行下一代测序更加容易。它也最适合大约 200 万次读取的小项目,尽管可以分解较大的项目,然后稍后合并结果。
可以在 Maqview 或平板电脑中查看对齐结果。
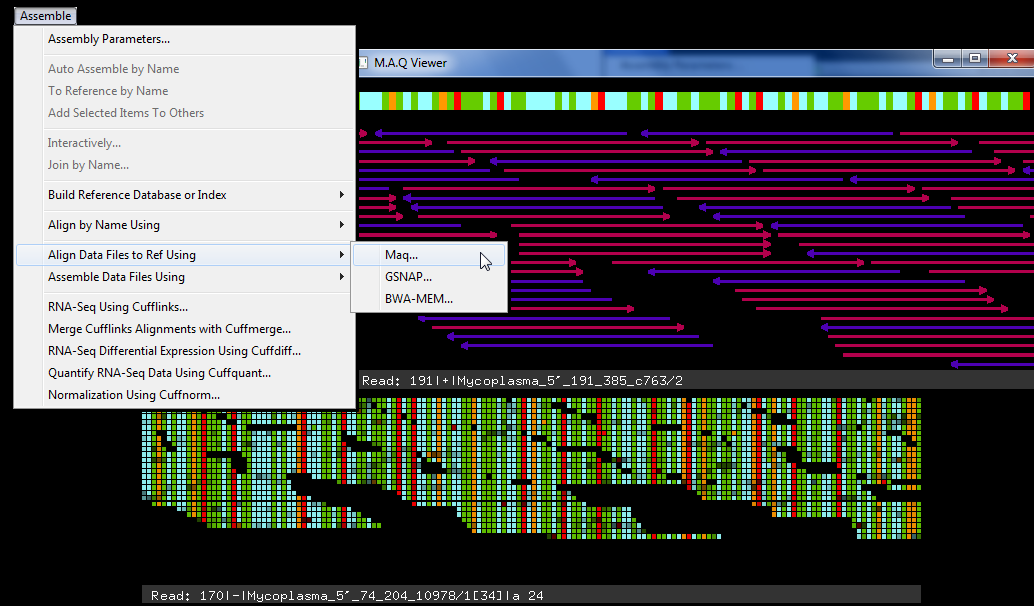
最初,Maq 被写入运行的命令行。Sequencher 提供了一个简单易用的接口,它保护您免受命令行的利用,并且不必学习如何使用命令行参数。
如果您想了解有关下一代装配体的更多内容,请查看下一代序列对齐和高级下一代序列对齐教程。
(1)恒李对齐序列读取,克隆序列和装配序号与BWA-MEM 2013 arXiv:1303.3997v2+q-bio。GN+ 请注意,此引用是一个预印arXiv.org。
(2)Thomas D. Wu 和 Serban Nacu 快速和 Snp 容度检测复杂变体和拼接在短读生物信息学 2010 26: 873-881
(3)李恒、朱如安和理查德·德宾映射短DNA测序读取和调用变体使用映射质量得分基因组研究 18:1851-1858
您已对它进行排序,现在您已对齐它。接下来,检查使用新变体调用功能的变体的对齐方式。无论您是将读取与参考引导对齐器之一对齐,还是在其他地方采购对齐的 SAM/BAM 文件,您仍可以使用使用 SAMtools 的变体调用来检查变体。
此分析可用于在 SNP 容差对齐、甲基化容差对齐或 GSNAP 中新的 RNA A 到 I 编辑容差模式之后。然后将由此产生的 VCF 文件和 SAM 文件加载到您最喜爱的浏览器中,如果您没有试用 Tablet , 这是我们 DNA-Seq Tools 分发的一部分。为平板电脑提供 GTF 文件进行注释,您还可以查看功能中的变体。
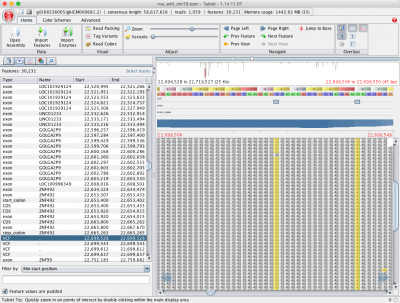
有关使用 SAMtools 进行变体呼叫的详细信息,请查看我们的高级下一代序列对齐教程。
Sequencher 5.2 和后使用多路复用方法,使用基于 BWA、GSNAP 或 Velvet 的基于参考的程序集的多路复用方法。多路复用是快速和容易的。每个DNA样本都有与之相关的特定条形码。测序器可以读取这些条形码,并划分数据到各种文件或”垃圾箱”。然后,排序器将采用每个”bin”中的序列,并执行装配或对齐。排序器的多路复用 ID 可与单端或配对端数据一起使用。对于配对端数据,对中每个读取的条形码必须在 5′ 端具有相同的条形码才能识别为对。您可以使用 Tablet 查看器查看多路复用程序集或对齐的结果。

有关多路复用 ID 的信息,请查看包含 BWA、GSNAP 和Velvet 教程的多路复用 ID。
筛选下一代测序产生的数百万读数在搜索候选 SNPs 时面临重大挑战。Maq 和 GSNAP 算法都包括 SNP 筛选功能。
Maq(1) 具有两个级别筛选过程,最初可搜索读取和参考序列之间的差异。接下来将执行筛选步骤,筛选初始结果,查找每列读取的相同类的最小数量变体,以及嵌入在高质量区域中的变体。研究结果在一份综合报告中提出。您还可以在 Maqview 或平板电脑中查看结果。
使用 GSNAP(2),SNP 分析采用不同的方法,查看以前报告的 SNP 和新候选项。用户必须提供已知 SNP 的列表以及读取和引用序列。GSNAP 对所有主要和次要等位基因执行可耐数的 SNP 对齐。该算法使次要等位基因能够与不匹配进行区分。研究结果在一份综合报告中提出。您还可以在平板电脑中查看结果。
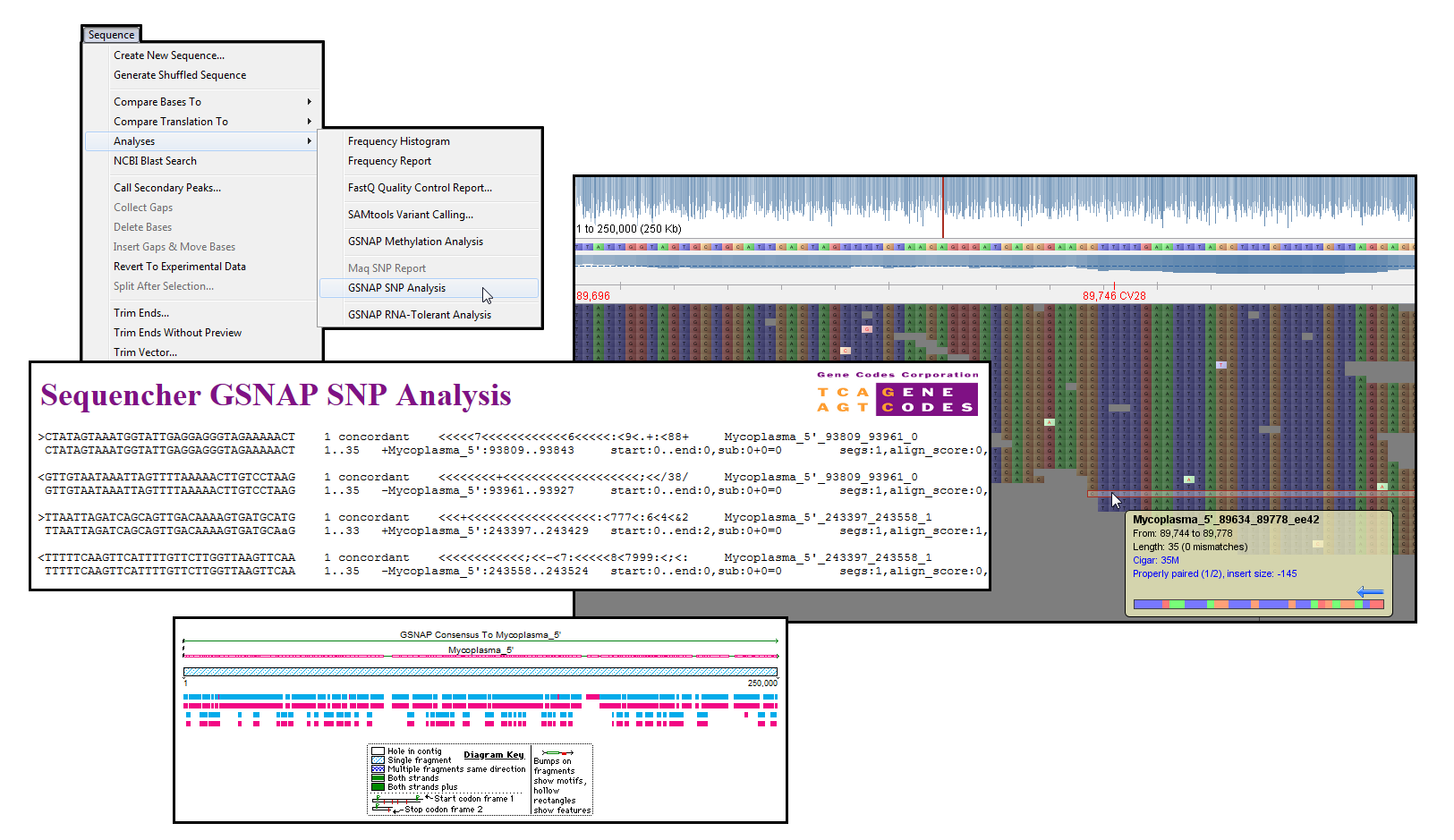
(1) 李恒、朱如安和理查德·
德宾使用测绘质量得分
进行短DNA测序读取和调用变体 基因组研究 18:1851-1858
(2) Thomas D. Wu 和
Serban Nacu 快速和 SNP 容检测复杂变体和
拼接在短读生物信息学 2010 26: 873-881
几十年来,DNA甲基化一直被认为在基因表达中起着重要的作用。当DNA用双硫化物处理时,任何细胞素都转化为葡萄干,除非细胞氨酸被甲基化。GSNAP(1) 将从双硫化处理 DNA 测序取取的下一代测序读取与参考序列对齐,以不掩盖真正不匹配的方式对齐它们,但仍使 C 与 T 不匹配,当未甲基化 DNA 使用双硫化 DNA 处理时发生。
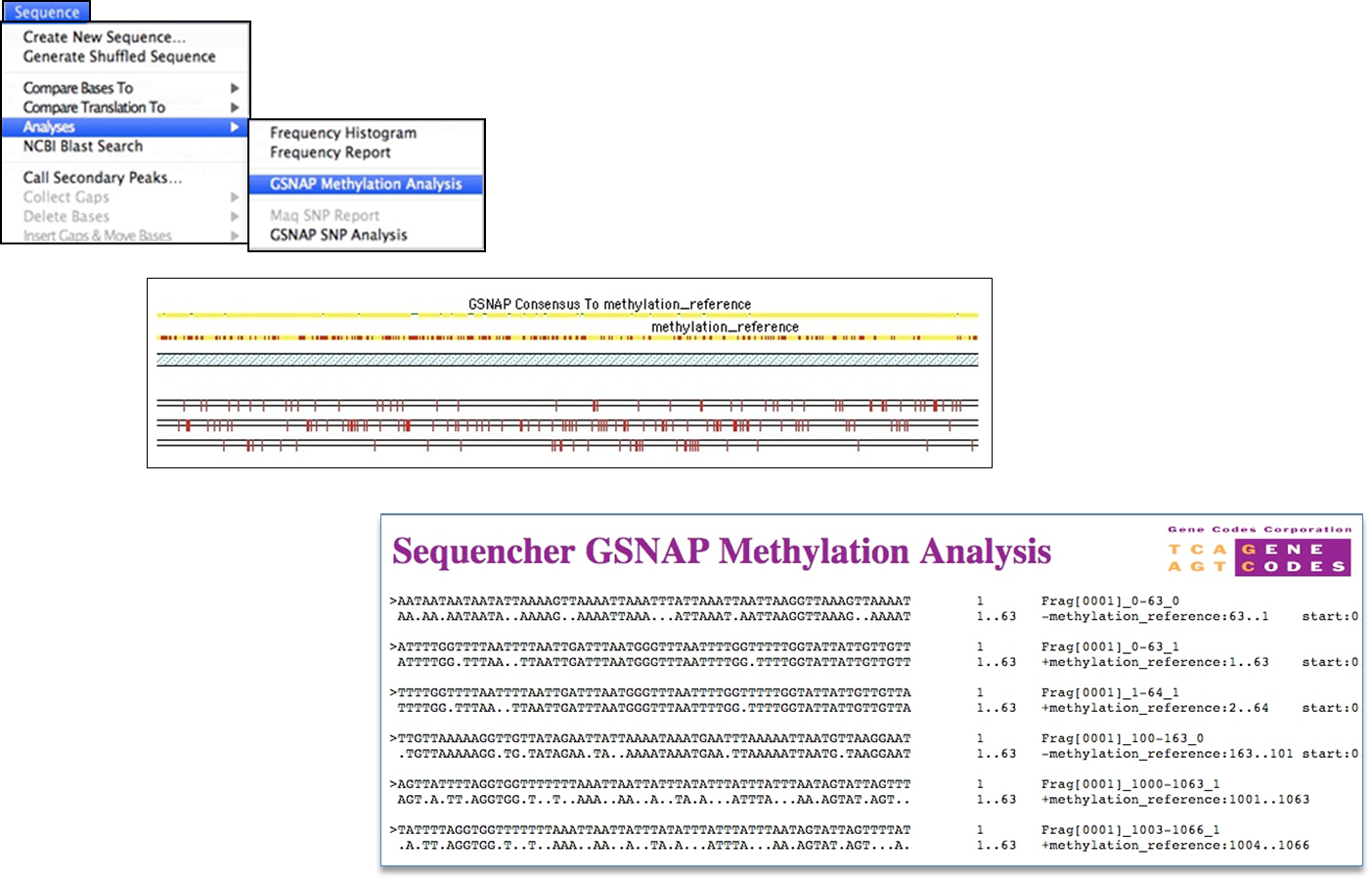
(1) Thomas D. Wu 和
Serban Nacu 快速和 SNP 容检测复杂变体和
拼接在短读生物信息学 2010 26: 873-881
GSNAP 已更新,现在包括 A 到 G 容差对齐模式。对于您的 mRNA 可能由 ADAR 基因编辑导致腺苷转化为内诺辛的情况,使用此模式。I 在排序时被视为 G。
此版本还引入了新的RNA耐受模式和甲基化分析的搁浅和非绞合模式。当您的实验室协议允许 5′ 到 3′ 基因组读取及其在每一条链上的反向补充时,请使用非绞合模式。
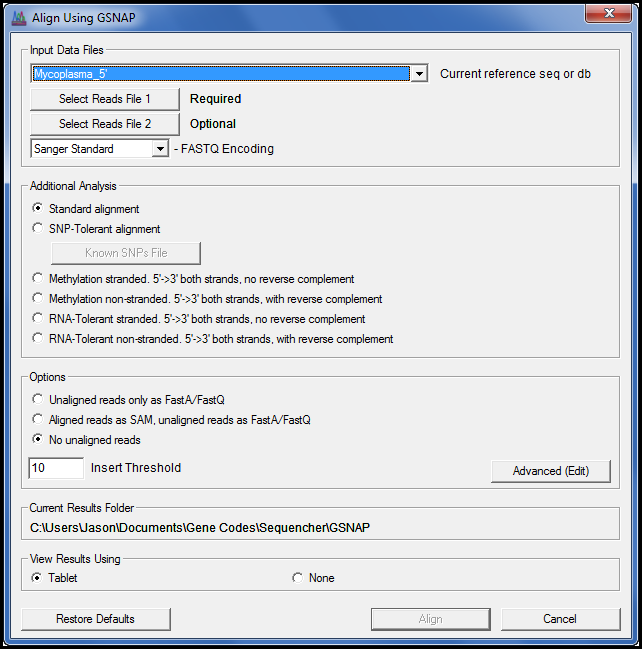
有关RNA容差对齐的信息,请查看高级下一代序列对齐教程。
Tablet(1) 是下一代序列对齐的高性能查看器。查看和查看您的 Maq、GSNAP、BWA-MEM 或 Velvet 结果,以许多不同的模式突出显示不同颜色的基座并读取方向。获取有关单个读取和读取对的信息,并排列读取的堆叠以显示对。平板电脑具有支持翻译、放大和缩小以及突出显示变体基础的控件,因此可以让您浏览您的阅读内容。
.
(1) 伊恩·米尔恩,米查·拜尔,琳达·卡德尔,保罗·肖,戈登·斯蒂芬,弗兰克·赖特和大卫·马歇尔平板电脑下一代序列组装可视化生物信息学 2010 26:40-402
您可以定义窗口的默认位置,而 Sequencher 会记住格式和共识选项的设置。您还可以保存所有设置,包括参考序列,作为项目模板,可以重复使用,以节省时间的分析设置!


“查找氨基酸”命令允许快速识别数据中的单个或氨基酸序列。使用单字母代码,插入您想找到的氨基酸,测序器将找到并突出显示这些氨基酸编码序列中的碱基。通过选择”查找氨基酸”窗口中的”查找下一步”按钮,测序器可以轻松转到符合搜索的下一组基础。
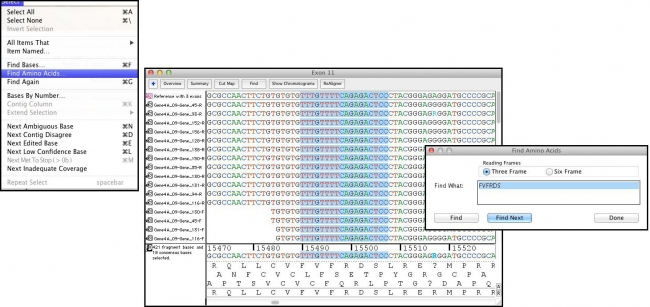
有关详细信息,请查看”测序用户手册”中的”查找氨基酸”命令。
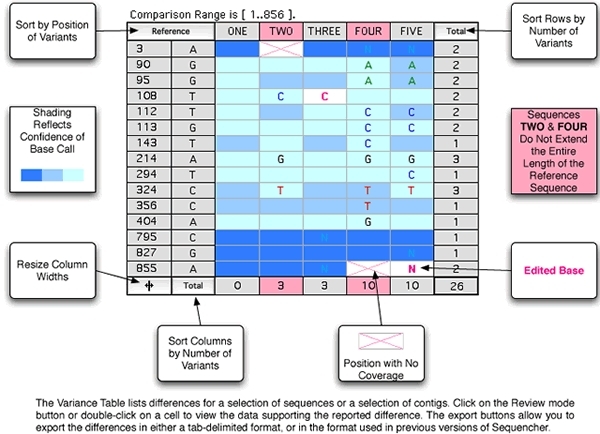
现在有一种非常简单的方法来比较DNA序列或连续共识序列,并发现这些序列之间的所有差异,只需几个击键。方差表允许您筛选大量序列数据,以便快速可视化您最感兴趣的基础。方差表中的每个单元格都链接到其原始数据,因此您可以轻松地直接验证或编辑表中的每个假设差异,并自动更新序列和色谱图数据。找到所有差异后,Sequencher 会为您提供各种导出和报告选项。报告页面中更详细地介绍了这些内容。
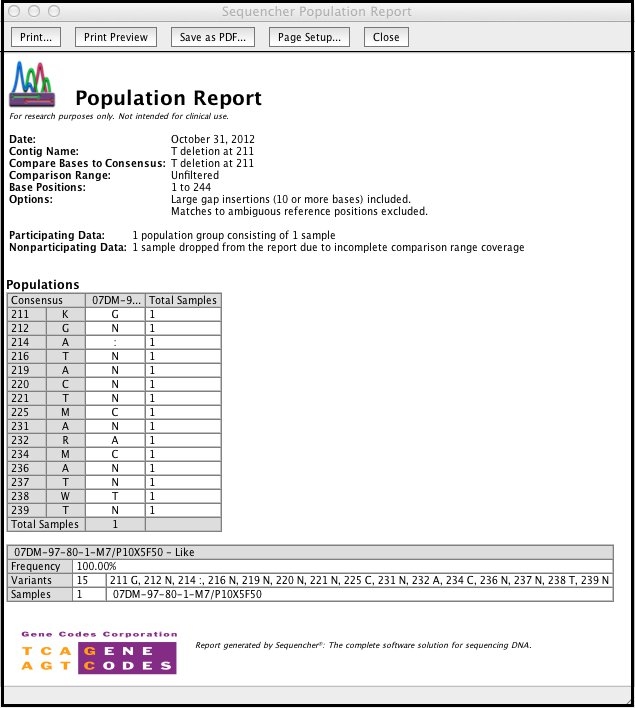
排序器的输出报告创建数据的可打印版本。这些报告提供分析工具,例如在人口报告中对等样本进行聚类。
人口报告是差异表中数据的唯一摘要。每个人口报告由两种类型的表组成;一个”人口”表和几个描述”人口”表中每个组的单个详细信息表。
“差异详细信息”报表提供排序器中用于支持差异表的所有信息。与”单个差异报告”中一样,摘要表列出了每个样本或列的变体。此外,在”差异详细信息”报表中,每个描述的变体的变体表遵循”详细信息表”。此表中的详细信息包括导致调用变体的任何数据的序列名称、方向和基本调用。如果有置信度或色谱图数据可用,则表中每个序列也包含此数据。色谱图数据包括次峰贡献的具体细节和色谱图的图像,每个变体最多有六个微量,尽管每个变体的微量跟踪数是可配置的。

排序连接表示对序列或序列组执行多重分析的新方法。连接通过允许您设置”通道”来扩展 Sequencher 的力量,这些通道可以运行使用不同的参数或数据库的不同分析。通过同时对不同通道上的多个序列或序列组运行分析,Sequencher 连接可以提高获取结果的速度。
测序连接现在更加灵活和可定制。可以重新打开现有连接会话,并为此添加新的序列。可以重命名连接会话以反映您的项目。现在有更多的选项用于将通道添加到连接会话,并且还可以命名每个通道。
您可以在连接会话中对单个序列执行 BLAST 搜索。可以设置通道来搜索具有不同参数或每个通道使用的数据库的 NCBI 数据库。可以设置本地 BLAST 通道来搜索计算机上的数据库。您还可以设置一个底向-BLAST 通道,以在感兴趣的区域上设计底转,并将其底转序列发送到您的 Sequencher 产品。底色序列将具有彩色底座,并应用底色特征。架构在单个窗口中显示来自多个通道的结果,以便您比较不同运行的结果。

您的连接会话将自动与项目一起保存。使用新的会话启动器,可以轻松创建新会话、向现有会话添加更多数据,或者只需查看和重新运行旧会话,以查看是否向之前查询过的数据库中添加了任何感兴趣的新序列。使用架构同时提供 BLAST、本地爆炸和底图-爆炸分析的图形概览。能够同时打开多个会话意味着您可以轻松查看所有结果。当您的结果最终从 NCBI 的网站过期时,您可以确保他们仍然会安全地在连接中。

您可以在连接会话中为一组序列运行肌肉对齐。结果可视为对齐序列或使用 Phylogram 进行可视化。可以设置不同的通道使用不同的参数。序列可以添加到现有会话中,以便不同的行查看不同的序列组。然后可以比较不同通道和行上的运行。

有关排序器连接的信息,请单击此处查看排序器连接教程。
BLAST 可能是您一直使用的工具,但有很多参数和选项,您可能没有每个承诺到内存的工具的影响。排序连接可帮助您在更少的时间中完成更多工作并比较结果。
是否要将结果与不同的设置进行比较?是否要将搜索分解为针对不同数据库的单个查询?也许最近发布了一些东西,现在又在数据库中,或者你了解一些关于你自己的数据的新东西。连接允许您一次运行许多不同的数据库搜索,返回结果的速度与完成一样快。然后,您可以保留所有结果,并使用架构进行比较。Schematic 是一个平行的图形显示,以 BLAST 无法完成的方式显示单个序列的所有分析结果。
如果您可以使用一个序列执行所有这些操作,请思考在同一会话中可以使用大量序列执行哪些操作。
如果您想了解有关BLAST和本地BLAST的更多信息,请查看 Sequencher 连接和本地BLAST tutorials。
使用底向-BLAST 分析序列并检查特异性。
并行分析 – 使用几组不同的设置,并在同一序列甚至不同的序列上同时运行,而无需编写脚本。
现在,只需单击一下即可将数据保存到项目中。
您的新底项会自动使用 GenBank 样式功能键进行标记,使其更易于在对齐中查看。底像按方向进行颜色编码,以便随时使用它们,并且由于它们的命名约定,您永远不会混淆它们。
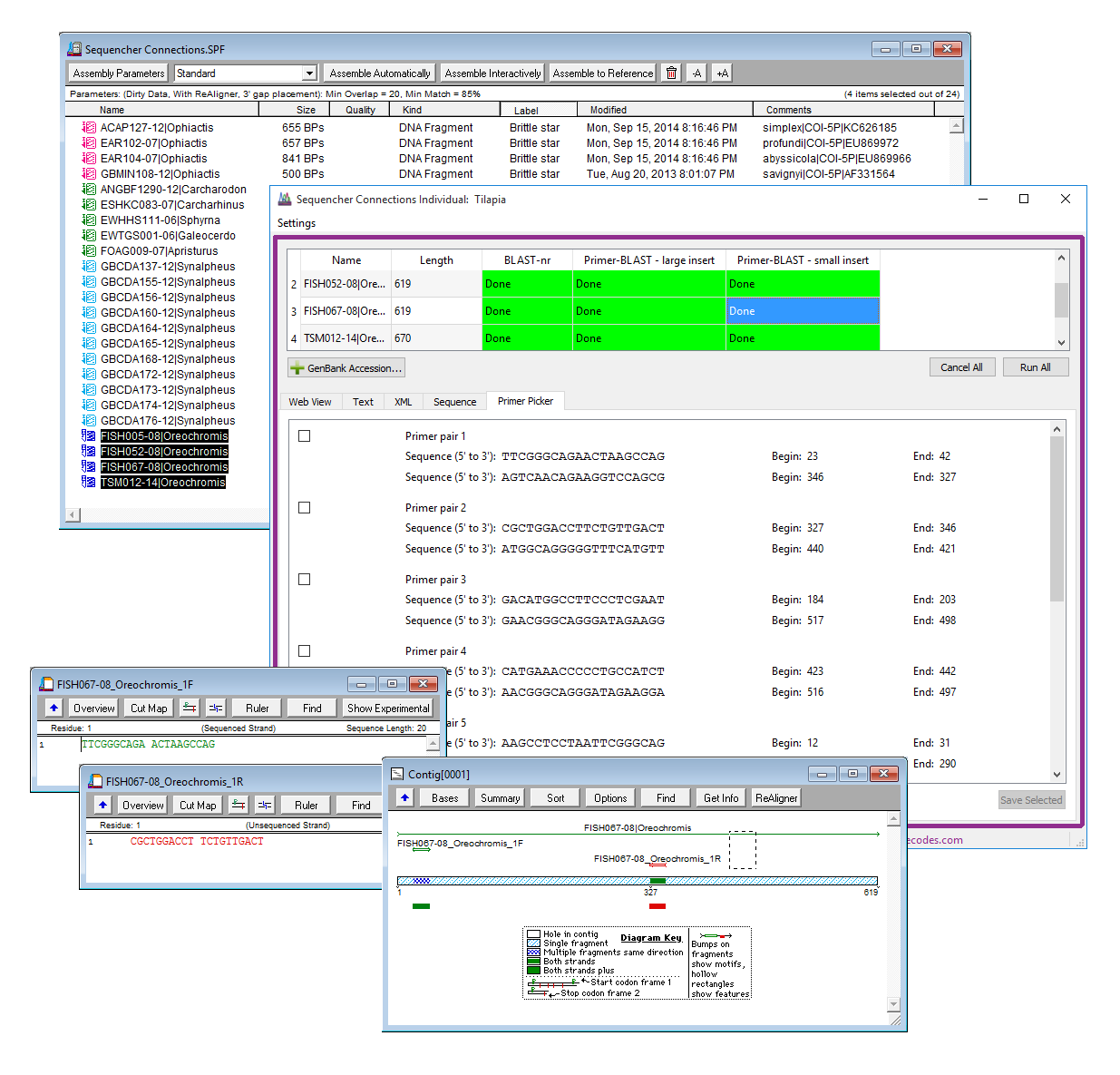
如果您想了解有关 Primer-BLAST 的更多信息,请查看排序器连接教程。
当您想要在 DNA 序列中查找保守区域时,请伸手查找肌肉。它比许多其他多序列对齐器速度快,精度更高。它易于使用,使用”排序连接”,您不仅可以看到对齐的区域,还可以以矩形或圆形树格式查看结果。
您可以在同一组序列上设置多个 MUSCLE 运行,每个分析(或通道)都设置为使用不同的参数。是否要将不同的数据集放在一起,但运行相同的 MUSCLE 参数?将它们添加到同一个连接会话,因为会话中的每一行可以包含一组不同的序列。MUSCLE 在不同的通道和行上运行,然后可以进行比较,从而为您提供数据的独特视角。